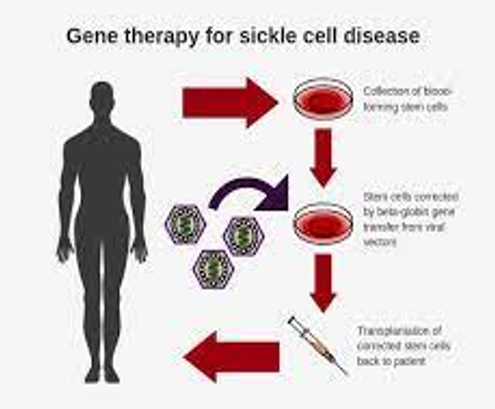
Sickle Cell Disease
Sickle Cell Disease
1. INHERITANCE
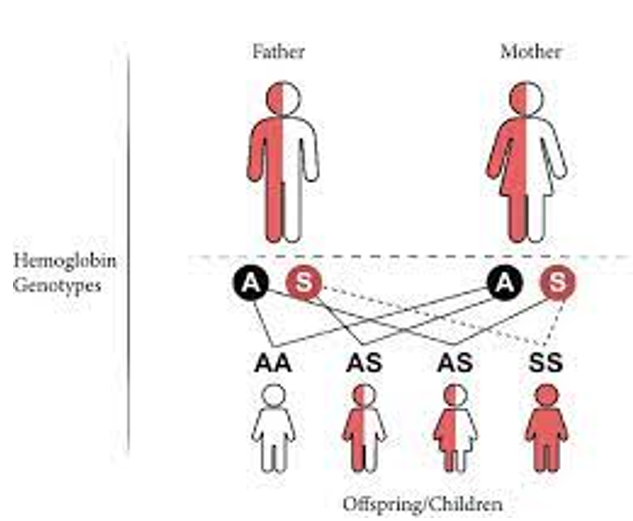
- Sickle cell disease is inherited in an autosomal recessive manner, meaning that both parents must carry the gene mutation for a child to be affected.
- Individuals with one normal hemoglobin gene (HbA) and one mutated gene (HbS) have a condition called sickle cell trait. They are carriers of the disease but usually do not show symptoms.
2. SYMPTOMS
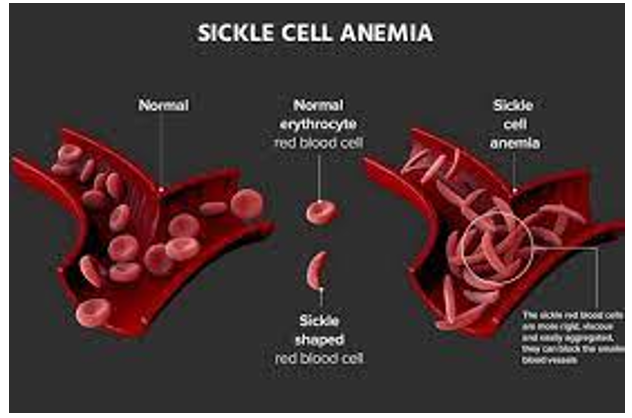
- SCD can cause various symptoms and complications, including pain crises (sudden, severe pain), anemia, fatigue, jaundice, organ damage, and increased susceptibility to infections.
- Pain crises occur when sickle-shaped red blood cells block blood flow, leading to pain and tissue damage.
3. DIAGNOSIS
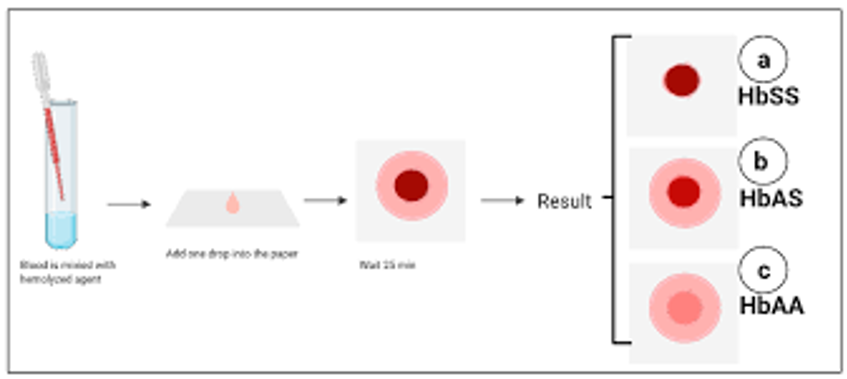
SCD is typically diagnosed through newborn screening tests or genetic testing. It can also be diagnosed before birth through prenatal testing.
4. MANAGEMENT
Medication – Management often involves medications to relieve pain, prevent infections, and manage SCD complications.
- Blood Transfusion – In severe cases, regular blood transfusions may be necessary to improve oxygen supply to the body’s tissues.
- Hydroxyurea – This medication helps increase the production of fetal haemoglobin, which reduces the formation of sickle-shaped cells.
- Bone Marrow Transplant – In some cases, a bone marrow transplant may be considered as a potential cure, especially in young patients with a compatible donor.

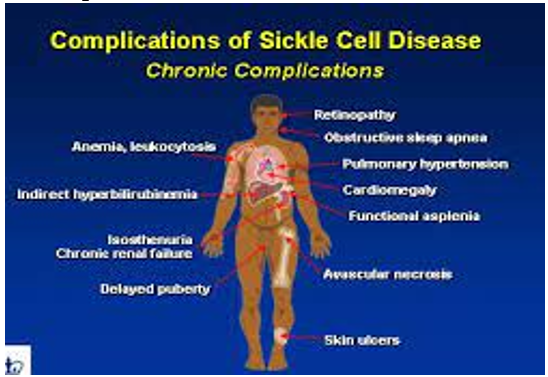
5. COMPLICATIONS
SCD can lead to various complications, including stroke, acute chest syndrome (a condition similar to pneumonia), organ damage, and impaired growth in children.
6. PREVENTION
The best way to prevent sickle cell disease is through genetic counseling and testing. Carriers can make informed decisions about family planning with the help of genetic counseling.
Prevention of sickle cell crisis is also important and this includes: Rehydration (fluid intake), correct dosage of hydroxyurea and other medication, avoiding extreme weather condition effects by keeping warm and normal temperature, balanced diet rich in iron and folate like green leafy vegetables, fruits, dairy products.
7. RESEARCH & DEVELOPMENT
Ongoing research aims to find new treatments and potential cures for SCD. Raising awareness about the disease is crucial to promote early diagnosis, access to proper healthcare, and improved quality of life for individuals living with SCD.
It’s important to note that the management and prognosis of SCD vary widely based on the individual’s specific genotype, the severity of the disease, and the availability of healthcare resources. Early diagnosis, comprehensive care, and a supportive community play significant roles in improving the lives of individuals affected by sickle cell disease.